






British Medical Journal speaks of CRIMINALIZING QUESTIONING VACCINE SAFETY!!! (youtu.be)
your ignorance is showing .. ha ha ha
do you even bother reading before you post?
article says NOTHING about alleged safety of mRNA "vaccines" aka gene therapy that NO one in the world knows what long term side effects will be.
NO mRNA "vaccine" has passed phase 3 safety trials. simply not possible for a experimental drug only 9 months old. big pharma are NOT responsible for damages from side effects in America, then factor in US gov is NOT responsible damages from experimental "vaccines".
one very important factor that probably beyond your comprehension level. currently NO one in the world has access to a complete SARS COV-2 genome, only snippets. if NO one can get access to a complete genome, then how in the world can anyone measure how effective said mRNA "vaccine" gene therapy is?
Editorials
Developing a vaccine for covid-19
BMJ 2020; 369 doi: https://doi.org/10.1136/bmj.m1790 (Published 04 May 2020)Cite this as: BMJ 2020;369:m1790
Read our latest coverage of the coronavirus pandemic
Old and new strategies are being investigated in an unprecedented worldwide effort
- Sarah Caddy, Wellcome Trust clinical research career development fellow
The rapidly developing covid-19 epidemic has stimulated an enormous effort to develop vaccines against the coronavirus SARS-CoV-2. At least six vaccine candidates have entered clinical trials across the globe, with more than 80 other candidates reported to be in preclinical stages.1 This means many different approaches are being moved forward at the same time. However, the road to successful vaccine licensure is treacherous, and only a handful of these vaccines may make it.
No vaccines are currently licensed for any of the other coronaviruses affecting humans—SARS-CoV-1, MERS-CoV, and minor cold viruses. Economic reasons are undoubtedly a major factor for the absence of these vaccines, but vaccine design is also a challenge; immune responses to natural coronavirus infections can be short lived, and some trial vaccines for SARS-CoV-1 raised safety concerns in animal models.2 The development of a SARS-CoV-2 vaccine therefore may not be straightforward.
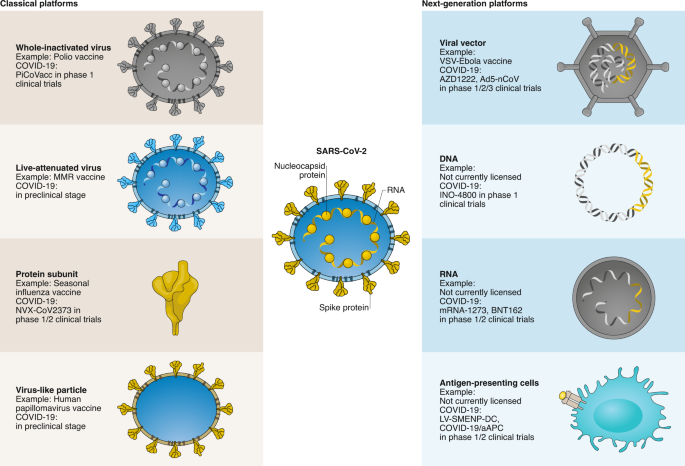
The multiple strategies to vaccine development for covid-19 include both traditional methods and next generation techniques. Historically, vaccines comprised inactivated whole virus, attenuated virus (less virulent but still immunogenic), or parts or subunits of the virus. Live vaccines are not likely to be attempted for covid-19 for safety reasons, but an inactivated whole virus vaccine has been taken through to preclinical trials in primates.3 When challenged with SARS-CoV-2, vaccinated macaques were protected from severe disease and cleared the virus within a week, whereas macaques receiving placebo developed severe interstitial pneumonia. A phase I-II human trial of this inactivated vaccine is now underway in China.4
Spike protein
Many other efforts are currently focused on the spike protein in SARS-CoV-2. This protein is part of the outer layer of the virus and is critical for entry into cells. Antibodies that target the spike protein can block virus entry, potentially inhibiting subsequent virus replication.5 The genetic sequence of the spike protein was released internationally on 10 January 2020, providing a blueprint for vaccine development.67
Widely reported UK contributions towards a SARS-CoV-2 vaccine are based on the spike protein. Scientists at the University of Oxford have modified a chimp adenovirus vector to carry the spike protein gene. When the adenovirus invades human cells, the spike protein will be produced, becoming a potential target for an immune response. The clinical trial for this vaccine started on 23 April and plans to recruit over 1000 volunteers.8
The use of messenger RNA as a vaccine is a relatively new strategy, and no licenced vaccines have yet used this method. The concept is simple though—inject mRNA coding for the spike protein and let the host make the protein. One advantage of this approach is a reasonably straightforward route to manufacture, allowing rapid scaling up of production. The first mRNA vaccine entered clinical trials in the US six weeks ago, and preliminary results are eagerly awaited. Related work is ongoing at Imperial College London, with promising results in mice released at the end of April.9
Other vaccine strategies under consideration include injecting DNA coding for the spike protein or the actual spike protein (“recombinant protein”). Others are using just the tip domain of the spike protein as this is the part that targets the receptors on human cells. Examples of these approaches are likely to enter phase I clinical trials this year.
Repurposing other vaccines
Repurposing vaccines to treat covid-19 is being considered as an alternative means of virus control. Hundreds of vaccines are licensed worldwide for non-coronavirus pathogens, and associations have been made between general vaccine uptake in a country and covid-19 severity.
The current frontrunner is the BCG vaccine, normally directed against tuberculosis. BCG vaccine can stimulate broad, innate components of the immune system, offering some protection against a range of diseases from influenza to bladder cancer.1011 Several studies have now proposed an epidemiological link between population BCG coverage and reduced covid-19 incidence at a country level.1213 Although several rebuttal studies have also been published, at least five clinical trials are now recruiting healthcare workers to investigate whether BCG protects them against covid-19.14
Other potential repurposed vaccines include the oral polio vaccine15 and the MMR vaccine.16 All these existing vaccines have the advantage that they can begin phase III trials immediately as safety (phase I) and immunogenicity (phase II) have already been established. However, evidence for their use must be regarded as tenuous at this point.
Which vaccine will make it successfully through clinical trials first? It’s too early to tell, and in an ideal world we would have several safe and effective vaccines. No single vaccine will be suitable for everyone, everywhere. Access will be particularly challenging for low income countries, where financial support will be essential.
Having choice will also increase the scale of overall production, using a variety of manufacturing options. While fast tracking research and development is an option in all well resourced countries, the most realistic time frame for the production at scale of any safe and effective vaccine against covid-19 still stands at over a year.
Acknowledgments
I thank Gordon Dougan for constructive comments.
Footnotes
References
- Competing interests: The BMJ has judged that there are no disqualifying financial ties to commercial companies. The authors declare no interests. The BMJ policy on financial interests is here: www.bmj.com/sites/default/files/attachments/resources/2016/03/16-current-bmj-education-coi-form.pdf.
- Provenance and peer review: Commissioned; not externally peer reviewed.
- ↵
Milken Institute. COVID-19 treatment and vaccine tracker. 2020. https://milkeninstitute.org/sites/default/files/2020-04/Covid19TrackerNEW4-21-20-2.pdf
- ↵
. SARS-CoV-2 vaccines: status report. Immunity2020;52:583-9.doi:10.1016/j.immuni.2020.03.007 pmid:32259480
- Amanat F,
- Krammer F
CrossRefPubMedGoogle Scholar- ↵
Gao AQ, Bao L, Mao H, et al. Rapid development of an inactivated vaccine for SARS-CoV-2. bioRxiv 2020.04.17.046375. [Preprint.] 2020. doi:10.1101/2020.04.17.046375.
Abstract/FREE Full TextGoogle Scholar- ↵
Safety and immunogenicity study of inactivated vaccine for prophylaxis of SARS CoV-2 infection (covid-19). Trial No NCT04352608. https://clinicaltrials.gov/ct2/show/NCT04352608.
- ↵
. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell2020;181:281-292.e6. doi:10.1016/j.cell.2020.02.058 pmid:32155444
- Walls AC,
- Park YJ,
- Tortorici MA,
- et al
CrossRefPubMedGoogle Scholar- ↵
. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature2020;579:270-3. doi:10.1038/s41586-020-2012-7 pmid:32015507
- Zhou P,
- Yang X-L,
- Wang X-G,
- et al
CrossRefPubMedGoogle Scholar- ↵
. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med2020;382:727-33. doi:10.1056/NEJMoa2001017 pmid:31978945
- Zhu N,
- Zhang D,
- Wang W,
- et al.,
- China Novel Coronavirus Investigating and Research Team
CrossRefPubMedGoogle Scholar- ↵
Oxford University. A study of a candidate COVID-19 vaccine (COV001). Trial No NCT04324606. https://www.clinicaltrials.gov/ct2/show/NCT04324606
- ↵
Mckay PF, Hu K, Blakney AK, et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine induces equivalent preclinical antibody titers and viral neutralization to recovered COVID-19 patients. bioRxiv 2020.04.22.055608 [Preprint.] 2020.doi:10.1101/2020.04.22.055608
Abstract/FREE Full TextGoogle Scholar- ↵
. Non-specific effects of BCG vaccine on viral infections. Clin Microbiol Infect2019;25:1473-8. doi:10.1016/j.cmi.2019.04.020 pmid:31055165
- Moorlag SJCFM,
- Arts RJW,
- van Crevel R,
- Netea MG
CrossRefPubMedGoogle Scholar- ↵
. Bacillus Calmette-Guérin (BCG) therapy for bladder cancer: an update. Immunotargets Ther2020;9:1-11. doi:10.2147/ITT.S202006 pmid:32104666
- Guallar-Garrido S,
- Julián E
CrossRefPubMedGoogle Scholar- ↵
Miller A, Reandelar MJ, Fasciglione K, et al. Correlation between universal BCG vaccination policy and reduced morbidity and mortality for COVID-19: an epidemiological study. MedRxiv 2020.03.24.20042937 [Preprint.] doi:10.1101/2020.03.24.20042937.
Abstract/FREE Full TextGoogle Scholar- ↵
Dayal D, Gupta S. Connecting BCG vaccination and COVID-19: additional data. MedRxiv 2020.04.07.20053272. [Preprint.] 2020, doi:10.1101/2020.04.07.20053272
Abstract/FREE Full TextGoogle Scholar- ↵
. https://www.clinicaltrials.gov/ct2/results?cond=COVID-19+&term=vaccine
- US National Library of Medicine
Google Scholar- ↵
Chumakov K, Gallo R. Could an old vaccine be a godsend for new coronavirus? 2020.https://eu.usatoday.com/story/opini...otential-treat-coronavirus-column/5162859002/
- ↵
Young A, Neumann B, Mendez RF, et al. Homologous protein domains in SARS-CoV-2 and measles, mumps and rubella viruses: preliminary evidence that MMR vaccine might provide protection against COVID-19. MedRxiv 2020.04.10.20053207. [Preprint.] 2020. doi:10.1101/2020.04.10.20053207
Abstract/FREE Full Text
your ignorance is showing .. ha ha ha
do you even bother reading before you post?
article says NOTHING about alleged safety of mRNA "vaccines" aka gene therapy that NO one in the world knows what long term side effects will be.
NO mRNA "vaccine" has passed phase 3 safety trials. simply not possible for a experimental drug only 9 months old. big pharma are NOT responsible for damages from side effects in America, then factor in US gov is NOT responsible damages from experimental "vaccines".
one very important factor that probably beyond your comprehension level. currently NO one in the world has access to a complete SARS COV-2 genome, only snippets. if NO one can get access to a complete genome, then how in the world can anyone measure how effective said mRNA "vaccine" gene therapy is?


 QUICK TAKEEfficacy and Safety of mRNA-1273 SARS-CoV-2 Vaccine 02:31
QUICK TAKEEfficacy and Safety of mRNA-1273 SARS-CoV-2 Vaccine 02:31






 QUICK TAKESafety and Efficacy of the BNT162b2 Covid-19 Vaccine 03:00
QUICK TAKESafety and Efficacy of the BNT162b2 Covid-19 Vaccine 03:00





